![$\displaystyle r=k[\ce{RuH4(PPh3)3}][\mathrm{keton}]$](img254.png) |
(13.2) |
Az 1970-es években a C=C kötés hidrogénezése során elért sikereket követően a kutatók figyelme kiterjedt a szén-heteroatom kettőskötések aktiválására is, és a ketonok alkoholokká történő redukálására irányuló kísérletek sikeresek voltak. Nem sokkal később Linn és Halpern górcső alá vette a ketonok hidrogénezését a ciklohexanon szubsztrátumon keresztül.Linn Jr and Halpern [1987] Feltételezték, hogy a RuH4(PPh3)3 képlettel jellemezhető komplex valójában egy RuH2(η2-H2)(PPh3)3 dihidrid-dihidrogén Ru(II) komplex. A szerzők a hidrogénezés sebességi egyenletére az alábbi összefüggést írták fel:
|
(13.2) |
![\includegraphics[width=0.9\textwidth]{hidrogen/rucocnhydrog}](img255.png)
|
Linn és Halpern erre a jelenségre azt a magyarázatot találta, hogy a lassú előegyensúly, azaz a H2 molekula/ligandum disszociációja (1) és az újbóli koordinációja (4) egymás ellenében hatnak és kioltják egymást. Mindez nem befolyásolja a keton szubsztrátumtól való elsőrendű függést.
A heteroatomot tartalmazó szubsztrátum a magános párján keresztül koordinálódik a ruténiumhoz (2). Keton esetén alkoxid közti termék keletkezik (3), majd következik a H2 koordinációja (4). Feltehetőleg 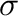 -metatézissel bekövetkezik az alkohol gyors eliminációja és egy telítetlen monohidrid képződik (5), ami aztán vagy felvesz egy újabb dihidrogént, vagy reagál a következő ketonnal.
-metatézissel bekövetkezik az alkohol gyors eliminációja és egy telítetlen monohidrid képződik (5), ami aztán vagy felvesz egy újabb dihidrogént, vagy reagál a következő ketonnal.
Az előbb látott esetben is, és jellemzően a foszfintartalmú katalizátoroknál a hidrid transzfer a fém közvetlen koordinációs övezetében megy végbe, így nevezhetjük ezt a folyamatot belső szférás hidrid transzfernek. Az 1990-es években Noyori kutatócsoportjával áttörést ért el az optikailag aktív alkoholok előállítása során, az általuk fém-ligandum kétfunkciós katalízisnek nevezett módszerrel. Először arra jöttek rá, hogy diaminok jelenléte legalább egy N-H csoporttal a Ru(II) koordinációs övezetében jelentős mértékben növelte a katalizátor aktivitását. Királis diaminok (például (R,R)-H2NCHPhCHPhNH2 (R,R)-dpen) és királis difoszfinok (pl. binap) egyidejű alkalmazásával viszont az aktivitás megőrzése mellett nagyobb optikai hozamot is kaptak. A jelenség magyarázata a külső szférás hidrid transzfer, melynél a karbonilvegyület szubsztrátum stabilizálásában az N-H kötésnek jelentős szerepe van (13.12 ábra).
Ha C=N kettőskötés hidrogénezéséről van szó, pl. Schiff-bázisok esetében, akkor az iridiumtartalmú rendszerek sokszor jó alternatívát jelentenek a Ru-tartalmú rendszerekhez képest. N-Aril-imineket több szerző is igen jó enantioszelektivitással hidrogénezett a megfelelő aminokká, jellemzően királis difoszfinokat tartalmazó Ir-komplexekkel.
A reakció mechanizmusát Osborn és Chan tanulmányozta különféle Ir-difoszfin-jodo komplexek előállításával és spektroszkópiai vizsgálatával. Arra a következtetésre jutottak, hogy prekurzortól függetlenül, hidrogén nyomás alatt a koordinatíve telítetlen Ir(III)-hidrid komplex az aktív katalizátor. A reakció mechanizmusát a 13.13 ábra mutatja be.Ng Cheong Chan and Osborn [1990]
![\includegraphics[width=0.65\textwidth]{hidrogen/irschiffhydrog}](img257.png)
|
Az iridiumos C=N kettőskötés hidrogénezés mechanizmusa eléggé eltér a ródiumos alkén hidrogénezéses mechanizmustól. Éredekes és meglehetősen ritka sajátsága ennek a rendszernek, hogy a ciklus során a köztitermékek végig Ir(III) komplexek, tehát nincsen benne oxidatív addíciós/reduktív eliminációs lépés.
A katalitikus ciklust az imin koordinációja indítja, mely 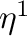 -N komplexet alkot a központi atommal (1). A következő lépésben az imin
-N komplexet alkot a központi atommal (1). A következő lépésben az imin 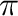 -koordinációra vált, így
-koordinációra vált, így  adduktok keletkeznek (2), melyek királis rendszer esetén diasztereomerikus viszonyban vannak egymással. A hidrid aztán átvándorol a (nitrogénhez kapcsolódó)
adduktok keletkeznek (2), melyek királis rendszer esetén diasztereomerikus viszonyban vannak egymással. A hidrid aztán átvándorol a (nitrogénhez kapcsolódó) 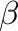 -szénatomra amido komplexet eredményezve (3). A záró lépésben egyidejűleg megy végbe -metatézissel az imin hidrogenolízise amin termékké, és a Ir-H kötés keletkezése, azaz a katalitikusan aktív komplex regenerálódása (4).
-szénatomra amido komplexet eredményezve (3). A záró lépésben egyidejűleg megy végbe -metatézissel az imin hidrogenolízise amin termékké, és a Ir-H kötés keletkezése, azaz a katalitikusan aktív komplex regenerálódása (4).
![\includegraphics[width=0.5\textwidth]{hidrogen/ruinnerouter}](img256.png)