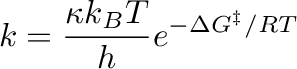![\includegraphics[width=0.9\textwidth]{kinetika/contour_map_path}](img53.png)
|
Mielőtt a matematikai/fizikai alapokat megtekintenénk, tegyünk gondolatban egy kirándulást egy dimbes-dombos vidéken, ahol A pontból el kell jutnunk B pontba. A kiindulási és célállomás adott, ezeken nem tudunk változtatni. Adott ezeknek a tengerszint feletti magassága is, tehát a szintkülönbség, amivel alacsonyabban (vagy magasabban) leszünk az út végén az tisztán attól függ, hogy honnan hová akarunk eljutni. Az viszont már tőlünk függ, hogy a több lehetséges útvonal közül, melyek völgyeken és hágókon át vezetnek, melyiket választjuk ki (4.1 ábra).
|
|
Ami a katalitikus reakciókat illeti, szinte pontosan ugyanez a helyzet, ha A-ból B-be kell eljutnunk, azaz adottak a kiindulási anyagok és adottak a termékek. Tengerszint feletti magasság helyett itt persze a szintvonalak közvetlenül jelentenek energia értékeket. Megjegyzendő, hogy a reakciútvonalak ilyesfajta ábrázolása a Born-Oppenheimer közelítés alkalmazásával képzelhető el, melynek lényege, hogy a molekulák kvantummechanikai leírásában elkülöníti az elektronok és az atommagok mozgását, mivel az atommagok tömege sokkal nagyobb, mint az elektronoké. Ennek köszönhetően az atommagok mozgása lassúbb, így az elektronok mozgását a kvázi-stacionárius atommagok körül vizsgálhatjuk. Ezáltal egy potenciálenergia-felületet (PES) kapunk, amely az atommagok különböző konfigurációihoz tartozó elektronenergiákat ábrázolja. A PES használatával egyszerűbbé válik a kémiai reakciók leírása, mivel a reakcióútvonalakat, a kiindulási anyagokat, a termékeket, a közti termékeket és az átmeneti állapotokat ezen a felületen ábrázolhatjuk, így részletesebb képet kapunk a reakciómechanizmusokról és az energiaprofilról. Természetesen a domborzati analógiánál a PES esetében némileg bonyolultabb a helyzet, ugyanis ott a tengerszint feletti magasságot ábrázoljuk a két másik helykoordináta függvényében, itt viszont az energiafüggvény változóinak száma mindenképpen több, mint kettő (hacsak nem kétatomos molekuláról van szó); ha 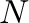 az atomok száma, akkor
az atomok száma, akkor 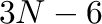 változós függvényünk van (lineáris molekulánál
változós függvényünk van (lineáris molekulánál 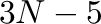 ). Az összes szabadsági fokból levonásra kerülnek a transzlációs és rotációs szabadsági fokok, azaz azok, amikor az összes atom egymáshoz képes egy irányba mozdul el, és azok is, amikor a háromdimenziós tér három tengelye körül forgatjuk el az összes atomot egyszerre, úgy hogy az egymáshoz viszonyított pozíciójuk nem változik. Így aztán a PES korrektebb elnevezése potenciálenergia hiperfelület, amit az Euklideszi térben értelmezünk.
). Az összes szabadsági fokból levonásra kerülnek a transzlációs és rotációs szabadsági fokok, azaz azok, amikor az összes atom egymáshoz képes egy irányba mozdul el, és azok is, amikor a háromdimenziós tér három tengelye körül forgatjuk el az összes atomot egyszerre, úgy hogy az egymáshoz viszonyított pozíciójuk nem változik. Így aztán a PES korrektebb elnevezése potenciálenergia hiperfelület, amit az Euklideszi térben értelmezünk.
Gyakorlati szempontból meglehetősen nehéz (sőt lehetetlen) lenne a reakcióútvonalakat háromnál több dimenzióban ábrázolni, ezért be szokták vezetni a reakciókoordináta fogalmát, mely a teljes útvonal kétdimenziós metszete. Itt már könnyedén meg lehet jeleníteni a potenciálenergia hiperfelület kiemelt pontjait, azaz az átmeneti állapotokat és a közti termékeket.
A termodinamikai egyensúly tehát adott, azaz a termékek és a kiindulási anyagok szabadentalpiái közötti különbség magának a reakciónak egy olyan alapvető tulajdonsága, amire nincs ráhatásunk. Körülbelül 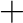 40 kJ/mol az a reakciószabadentalpia, aminél ha nagyobb a tervezett reakciónkra előzetesen kalkulált
40 kJ/mol az a reakciószabadentalpia, aminél ha nagyobb a tervezett reakciónkra előzetesen kalkulált 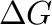 , akkor azon semmiféle katalizátor nem fog segíteni.4.1Ha a termodinamika nem húzza keresztül a számításunkat, akkor viszont arra van befolyásunk (persze sokszor csak elvben), hogy kiválasszuk a megfelelő katalizátort, azaz eldöntsük, hogy többféle lehetséges útvonal közül melyiket járjuk végig a kezdeti és a végállapot között.
, akkor azon semmiféle katalizátor nem fog segíteni.4.1Ha a termodinamika nem húzza keresztül a számításunkat, akkor viszont arra van befolyásunk (persze sokszor csak elvben), hogy kiválasszuk a megfelelő katalizátort, azaz eldöntsük, hogy többféle lehetséges útvonal közül melyiket járjuk végig a kezdeti és a végállapot között.
A kémiai reakció sebességi állandójának hőmérsékletfüggését az Arrhenius-egyenlet írja le. Az egyenlet alapvető összetevői az aktiválási energia, (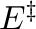 vagy néha
vagy néha 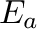 ), a preexponenciális faktor, (
), a preexponenciális faktor, (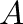 ), az egyetemes gázállandó, (
), az egyetemes gázállandó, (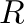 ), és az abszolút hőmérséklet (
), és az abszolút hőmérséklet (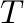 ). Az egyenlet azt mutatja, hogy a reakciósebességek nőnek a hőmérséklettel, ahogy a reagensek megkapják a sikeres reakcióhoz szükséges aktiválási energiát:
). Az egyenlet azt mutatja, hogy a reakciósebességek nőnek a hőmérséklettel, ahogy a reagensek megkapják a sikeres reakcióhoz szükséges aktiválási energiát:
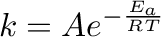 |
(4.1) |
preexponenciális faktor, mivel nincs egyértelmű fizikai definíciója. Ami a dimenzióját illeti, annak egyeznie kell a sebességi állandó dimenziójával (pl. 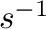 egy elsőrendű reakció esetén), mivel ez exponenciális tag dimenziómentes szám.
Megemlítendő, hogy nagyon kis aktiválási energiák esetén (kb. 20 kJ/mol alatt) már nem annyira a reakciómechanizmussal kapcsolatos kémiai folyamatok a meghatározók, hanem a részecskék közötti kölcsönhatás sebessége. Az ilyen eseteket diffúziókontrollált folyamatoknak nevezzük.
egy elsőrendű reakció esetén), mivel ez exponenciális tag dimenziómentes szám.
Megemlítendő, hogy nagyon kis aktiválási energiák esetén (kb. 20 kJ/mol alatt) már nem annyira a reakciómechanizmussal kapcsolatos kémiai folyamatok a meghatározók, hanem a részecskék közötti kölcsönhatás sebessége. Az ilyen eseteket diffúziókontrollált folyamatoknak nevezzük.
Az aktiválási paraméterek, mint az entalpia, entrópia és térfogat, kulcsfontosságúak a kinetika és a reakciómechanizmusok értelmezésében. Ezek a paraméterek az elemi reakciók esetében meghatározhatók, és segítenek a többlépcsős reakciók egyes lépéseinek elemzésében. Maga az aktiválás az a folyamat, amikor a reaktánsok szert tesznek arra az extra energiára, ami elegendő ahhoz, hogy el tudjanak jutni az átmeneti állapotig. Az energia nem egyenletesen oszlik el a részecskék között; egy részük energiája viszont meghaladja az aktiválási energiát. A statisztikus termodinamikában a Boltzmann eloszlás segítségével lehet kiszámolni az adott energiaszintet meghaladó populáció arányát. A Boltzmann eloszlás lényege a hőmérsékletet a nevezőben tartalmazó exponenciális tag, 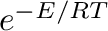 , ami hangsúlyos része az Arrhenius egyenletnek is.
, ami hangsúlyos része az Arrhenius egyenletnek is.
Az Eyring-egyenlet elsősorban a kémiai reakciók sebességállandójának hőmérsékletfüggését írja le. Ez egy alapvető fontosságú képlet a reakciókinetikában, mely arra szolgál, hogy meghatározza, hogyan változik egy reakció sebessége a hőmérséklet függvényében.
ahol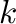 a sebességi állandó,
a sebességi állandó, 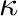 a transzmissziós koefficiens (értéke többnyire 1),
a transzmissziós koefficiens (értéke többnyire 1), 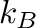 a Boltzmann állandó, a hőmérséklet, az egyetemes gázállandó,
a Boltzmann állandó, a hőmérséklet, az egyetemes gázállandó,
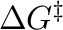 pedig az aktiválási szabadentalpia.
pedig az aktiválási szabadentalpia.
A képlet igen hasznosnak bizonyul a kémiai reakciók dinamikájának megértésében, és úgyszolván nélkülözhetetlen az aktiválási entalpia és az aktiválási entrópia mennyiségi meghatározásához. Ezen képessége miatt az Eyring-egyenlet széles körben alkalmazható problémás esetek megoldására is a reakciókinetikában, főként a reakciósebességek meghatározásán keresztül. Jól ismert az aktiválási paraméterek közti összefüggés:
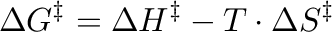 |
(4.3) |
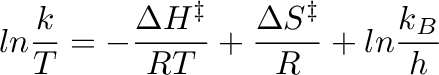 |
(4.4) |
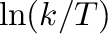 változót
változót 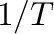 függvényében ábrázoljuk, akkor egy egyenest kapunk, melynek meredekségéből meghatározhatjuk az aktiválási entalpiát, a függőleges tengelyen kapott metszéspontból pedig az aktiválási entrópiát.
függvényében ábrázoljuk, akkor egy egyenest kapunk, melynek meredekségéből meghatározhatjuk az aktiválási entalpiát, a függőleges tengelyen kapott metszéspontból pedig az aktiválási entrópiát.